Salad Bowls & DNA Files: Mastering Fan-Out/Fan-In in the Cloud
- Melvin Jones
- Jun 9, 2025
- 2 min read


Building a Neighborhood Salad: Imagine you’re hosting a big picnic and decide to make one gigantic salad. You hand each of six neighbors a small bowl of lettuce and ask them to add their favorite topping - tomatoes, cucumbers, olives, croutons, carrots, or cheese - and then return their bowls to you. While they all chop and mix in parallel, you relax and catch up on other tasks. When every neighbor brings back their personalized bowl, you pour them together into a single, massive salad bowl and give it a gentle toss. This is fan-out / fan-in in action: send out the work to many helpers, then gather all the results into one delicious whole. In my solution, I break a human genome file (3.4GB) into smaller chunks and process them in parallel, then summarize their contents in a single file.
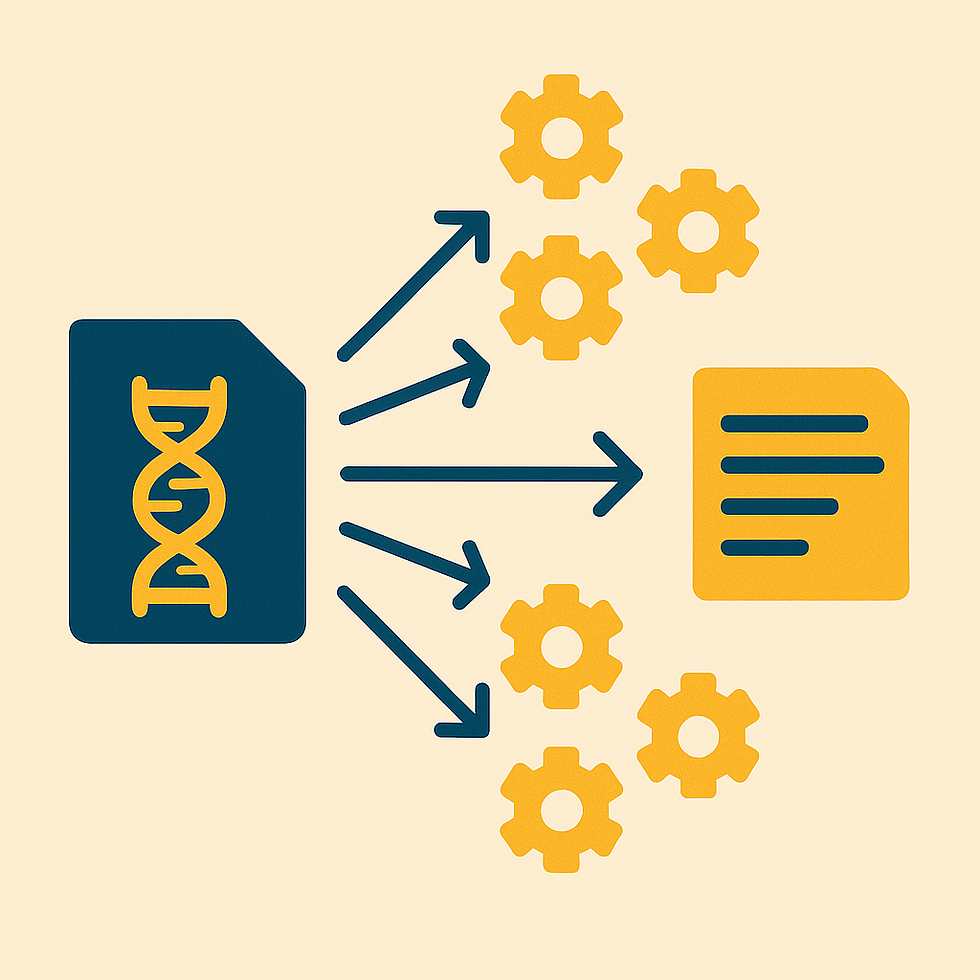
Tackling Gigantic Genome Files: In genomics research, a common problem is dealing with files so large that they are slow or impossible to process on a single machine. A human genome FASTA file can be tens or hundreds of gigabytes, making analyses like GC content calculation or k-mer counting painfully slow if done end-to-end. By slicing the file into manageable chunks, we can distribute the work across many parallel tasks in the cloud and complete the entire analysis in a fraction of the time.

High-Level Cloud Solution: My solution uses a simple API call to launch the workflow. A “starter” Lambda immediately queues up a “chunker” Lambda that splits the genome file in S3 into fixed-size pieces and then submits an AWS Batch array job on Fargate. Each Batch task picks up one chunk, runs a container that computes per-chunk metrics, and writes a small JSON summary back to S3. Once the last task finishes, an EventBridge rule triggers an “aggregator” Lambda that pulls in all the summaries, merges them into one master report, and writes the final JSON back to S3.
This pattern solves three core problems at once:
1) It turns a monolithic workflow into a set of independent, stateless tasks
2) It fully leverages on-demand cloud compute resources, so you only pay for what you useIt guarantees that if any chunk fails, you can retry that piece without reprocessing the entire genome.
3) The result is faster time to insight, more resilient pipelines, and a clear path to scaling up to even larger datasets. Other Real-World Examples: Beyond genomics, fan-out / fan-in shines wherever you have large, splittable workloads. For example, you might decode and transcode hours of raw video into multiple bitrate streams in parallel, then stitch them into a streaming package. Or you could crawl a huge list of web pages to extract product information, letting each child process run independently before you aggregate all price and availability data into a searchable catalog.
Try It Yourself: If you’d like to explore the full implementation, check out the genome-seq-batch-demo on my GitHub at https://github.com/mjones3/genome-seq-batch-demo. You’ll find Terraform templates, Lambda code, Docker files, and detailed instructions so you can run your own fan out fan in pipeline in minutes.



Comments